
Synthetic community [SynCom] transfer for the...

Find out more about the different routes to entry and our eligibility criteria
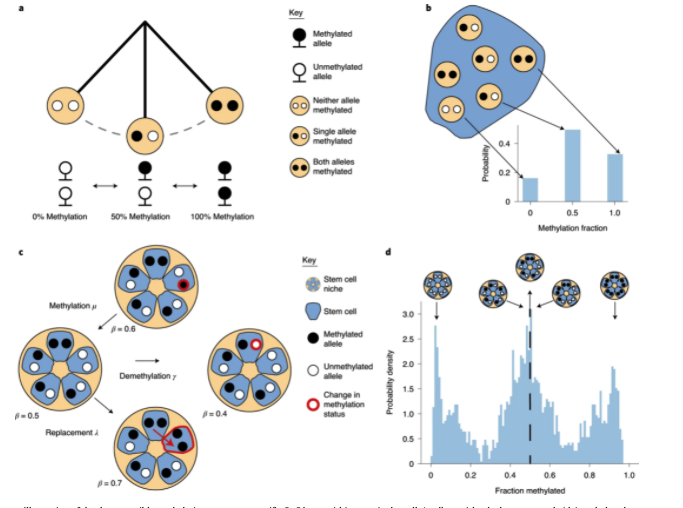
Here, we show that fluctuating DNA methylation marks can be used as clocks in cells where ongoing methylation and demethylation cause repeated ‘flip–flops’ between methylated and unmethylated states. We identify endogenous fluctuating CpG (fCpG) sites using standard methylation arrays and develop a mathematical model to quantitatively measure human adult stem cell dynamics from these data.
Small intestinal crypts were inferred to contain slightly more stem cells than the colon, with slower stem cell replacement in the small intestine. Germline APC mutation increased the number of replacements per crypt. In blood, we measured rapid expansion of acute leukemia and slower growth of chronic disease. Thus, the patterns of human somatic cell birth and death are measurable with fluctuating methylation clocks (FMCs).
The fates of individual human cells in vivo are difficult to reconstruct. In animal models, the use of transgenic or exogenous cell labeling enables straightforward clonal lineage tracing1,2,3,4,5,6,7,8,9,10, but in humans, these methods are precluded. Instead, human studies must use somatic genomic alterations, termed ‘molecular clocks’, to trace somatic cell fates. The key principle is that the ancestry of a population of cells is revealed by the somatic alterations shared amongst the cells: closely related cells are likely to share multiple alterations, whereas distantly related cells will have few alterations in common. Thus, human lineage tracing studies rely on the notion that the clonal history of a cell is recorded in its genome. Various types of somatic genomic alterations have been exploited for lineage tracing in human tissues, including mitochondrial DNA mutations11,12,13,14,15,16,17,18,19,20,21,22,23,24, DNA methylation at selectively neutral loci25,26,27,28,29,30,31,32, allelic loss at heterozygous loci33,34 and single-nucleotide variants detected by genome sequencing35,36,37,38,39,40,41,42,43,44,45,46,47.
Most molecular clocks use ‘unidirectional’ measurements that count the accumulation of changes since birth to infer the relatedness of lineages. The resolution at which a molecular clock can track clonal ancestry is a function of the rate at which genomic alterations accrue. A slow rate of alteration accrual can only reveal clonal dynamics occurring over long timescales. For example, genome sequencing studies of normal skin47, blood37, intestinal crypts38 and endometrial glands39 identified multiple subclones in each tissue, but in most cases, the reconstructed lineages diverged many years in the past, and recent cell turnover was not evident in the data. In comparison, a faster rate of alteration accrual has the potential to reveal rapid and/or recent clonal dynamics, but in practice, these approaches are compromised by ‘saturation’ wherein the same pattern of alterations evolve convergently in distinct clonal populations48, and effectively recording stops in childhood.
