
Synthetic community [SynCom] transfer for the...

Find out more about the different routes to entry and our eligibility criteria
In this study, we focused on understanding how interactions between CaMKIIα and the actin-crosslinking protein α-actinin-2 underlie long-lasting changes in dendritic spine architecture. We found that association of the two proteins was unexpectedly elevated within 2 minutes of NMDA receptor stimulation that triggers structural LTP in primary hippocampal neurons. Furthermore, disruption of interactions between the two proteins prevented the accumulation of enlarged mushroom-type dendritic spines following NMDA receptor activation. α-Actinin-2 binds to the regulatory segment of CaMKII. Calorimetry experiments, and a crystal structure of α-actinin-2 EF hands 3 and 4 in complex with the CaMKII regulatory segment, indicate that the regulatory segment of autoinhibited CaMKII is not fully accessible to α-actinin-2. Pull-down experiments show that occupation of the CaMKII substrate-binding groove by GluN2B markedly increases α-actinin-2 access to the CaMKII regulatory segment.
Furthermore, in situ labelling experiments are consistent with the notion that recruitment of CaMKII to NMDA receptors contributes to elevated interactions between the kinase and α-actinin-2 during structural LTP. Overall, our study provides new mechanistic insight into the molecular basis of structural LTP and reveals an added layer of sophistication to the function of CaMKII.
Changes in synaptic connections between neurons are fundamental to learning and memory (Citri and Malenka, 2008). Calmodulin-dependent protein kinase II (CaMKII) plays a central role in long-term potentiation (LTP) of excitatory synapses following influxes of Ca2+ into postsynaptic spines (Hell, 2014). Activation of CaMKII by Ca2+/CaM leads to phosphorylation of postsynaptic proteins including AMPA-type glutamate receptors which has the overall effect of increasing postsynaptic responsiveness to glutamate release (Anggono and Huganir, 2012). CaMKII also serves a structural function in LTP (Hojjati et al., 2007) by nucleating networks of protein-protein interactions through its modular oligomeric structure (Bhattacharyya et al., 2020), consistent with its extremely high abundance in dendritic spines (Erondu and Kennedy, 1985). CaMKII holoenzymes form through oligomerisation of the C-terminal hub domains of 12 subunits (Chao et al., 2011; Myers et al., 2017). The hub domains assemble into a two-tiered central ring from which the N-terminal kinase domains radiate (Chao et al., 2011; Myers et al., 2017).
Binding of Ca2+/CaM to a central regulatory segment releases the segment from the kinase domain enabling access to substrates and interaction partners (Yasuda et al., 2022). The CaMKII kinase domain has many documented substrates and binding partners (Özden et al., 2020). Formation of a highly stable complex between the CaMKII kinase domain and the C-terminal tail of NMDA receptor (NMDAR) GluN2B subunits is thought to be critically important for learning and memory (Sanhueza et al., 2011). Unlike the promiscuous kinase domain, the regulatory segment of CaMKIIα interacts only with α-actinins (Dhavan et al., 2002; Walikonis et al., 2001) besides CaM, while the sole binding partner of the hub domain is densin-180 (Strack et al., 2000b). Understanding how and when the regulatory and hub regions of CaMKII engage in these interactions is essential for a complete understanding of the structural role of CaMKII in LTP (Hell, 2014).
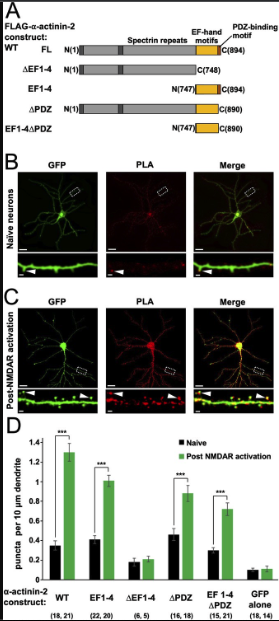
α-Actinins are structural proteins that form antiparallel rod-like dimers (de Ribeiro et al., 2014). In the heart, they are a key component of Z-discs where they crosslink actin filaments to titin (Young et al., 1998). In dendritic spines, α-actinins serve a more complex function with many additional binding partners including CaMKII, NMDARs (Wyszynski et al., 1997), PSD-95 (Matt et al., 2018), CaV1.2s (Hall et al., 2013), and densin-180 (Walikonis et al., 2001). Three of the four α-actinin isoforms have been detected in the postsynaptic density (Walikonis et al., 2000). The most abundant is α-actinin-2, which localises to dendritic spines of excitatory synapses (Rao et al., 1998; Wyszynski et al., 1998) where it interacts with the CaMKII regulatory segment independent of Ca2+ through its third and fourth EF hands close to its C-terminus (gold, Figure 1A; Jalan-Sakrikar et al., 2012). Association with actinin only weakly stimulates the activity of CaMKII towards certain substrates (Jalan-Sakrikar et al., 2012; Robison et al., 2005a), therefore the interaction is thought to serve a structural role. Immunogold labelling shows that α-actinin-2 is present within the postsynaptic density (Wyszynski et al., 1998). Knockdown or overexpression of α-actinin-2 in cultured hippocampal neurons leads to defects in spine formation, with α-actinin-2 knockdown breaking the link between NMDAR activation and spine enlargement (Hodges et al., 2014) – a process known as structural LTP.
